Inherited Retinal Degenerations Masquerading as Age-related Macular Degeneration in Older Adults
Authors
Kirk AJ Stephenson MD FRCSI*, Andrew W Kirker MD FRCSC, Cheryl Y Gregory-Evans PhD, Kevin Gregory-Evans MD FRCSC, Kaivon Pakzad-Vaezi MD FRCSC
Department of Ophthalmology & Visual Sciences, University of British Columbia.
DOI: https://doi.org/10.62856/djcro.v1i1.24.
*Corresponding author:
Kirk AJ Stephenson MD FRCSI
Section C
2550 Willow Street
Vancouver, B.C.
Canada, V5Z 0A6
E-mail: kirkstephenson@hotmail.com
Introduction
Inherited retinal degenerations (IRD) are the leading cause of blindness in the working-age population; however, those >75 years old represent up to 10% of all IRD patients.1-3 These conditions are heritable though de novo cases are frequent (~14%).4 Definitive treatments are lacking, particularly for advanced atrophic disease, though significant progress has been made with the first US FDA-approved subretinal gene therapy for biallelic RPE65-mediated retinopathy (Luxturna, Novartis).5 Though conditions such as age-related macular degeneration (AMD) explain a large proportion (53%) of macular visual loss in people >75 years, other conditions including medication toxicity (e.g., hydroxychloroquine, pentosan polysulfate), high myopia, and IRD must be excluded.6, 7
Herein, we describe two examples of elderly patients with a referring diagnosis of macular degeneration who were found to have a more complex genetic etiology.
Case One Report
An 82-year-old female was referred with atypical AMD. She had noticed bilateral reduced visual acuity for the past 10 years having ceased driving as she was aware of scotomata. She reported no night blindness or peripheral visual field constriction. She had a history of controlled hypertension and denied use of hydroxychloroquine or pentosan polysulfate. Her mother was diagnosed with AMD in her 80s but there was no other known family history of retinal disease.
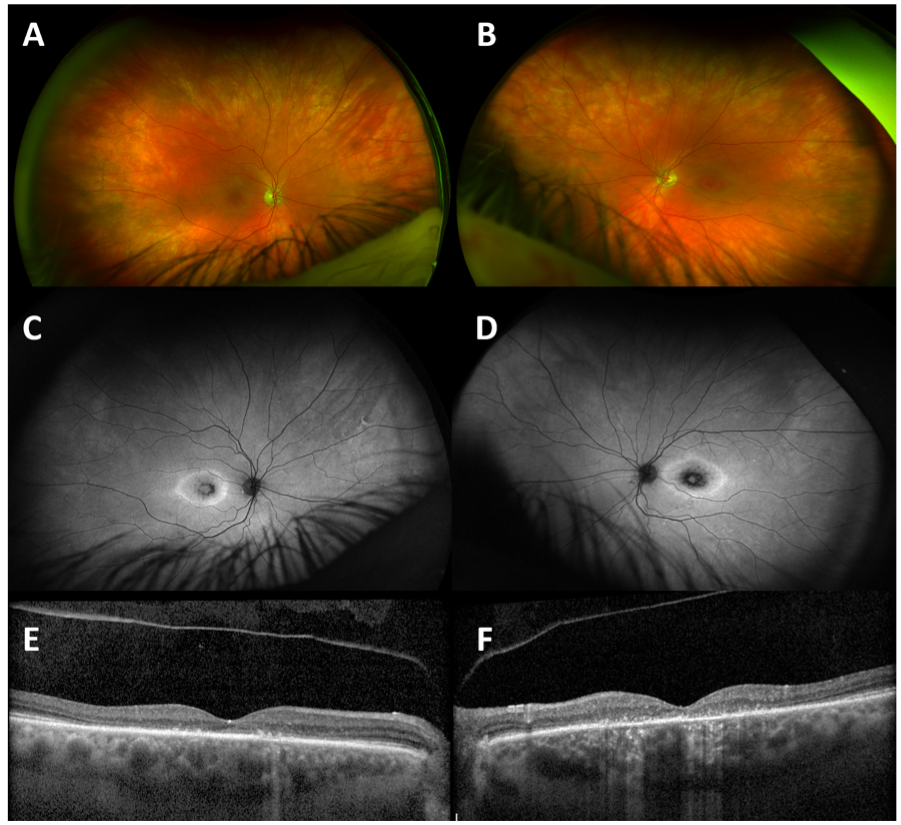
On examination, visual acuity (VA) was 20/60 in the right eye and 20/200 in the left eye. Intraocular pressures (IOP) were within the normal range, and she was bilaterally pseudophakic with posterior vitreous detachments in each eye. There was no evidence of intraocular inflammation, and media were clear. She had asymmetric bulls-eye maculopathy more marked in the left eye (Fig 1A, B). Autofluorescence imaging (Fig 1C, D) demonstrated disease at the level of the retinal pigment epithelium, and optical coherence tomography (OCT, Fig 1E,bF) confirmed central macular ellipsoid zone (EZ) loss, focal areas of increased signal transmission, and pachychoroid changes without macular edema or subretinal fluid.
The suspicion of a genetic etiology, despite presentation in late adulthood, prompted panel-based genetic testing for IRD-associated genes (Invitae Corp, Inherited Retinal Disorders Panel, 330 genes) revealing a heterozygous deletion of the entire CRX gene consistent with autosomal dominant (AD) cone-rod dystrophy (OMIM #120970).
Case Two Report
An unrelated 72-year-old male was referred with a central retinal vein occlusion (CRVO) in the left eye with a 2-week history of sudden monocular blurred vision and distortion, with an asymptomatic fellow eye. He had a previous ophthalmic diagnosis of geographic atrophy (GA) sparing his fovea in the right eye as well as bilateral glaucoma treated with topical dorzolamide/timolol drops. He was otherwise healthy taking only oral aspirin 81mg. There was no family history of eye disease.
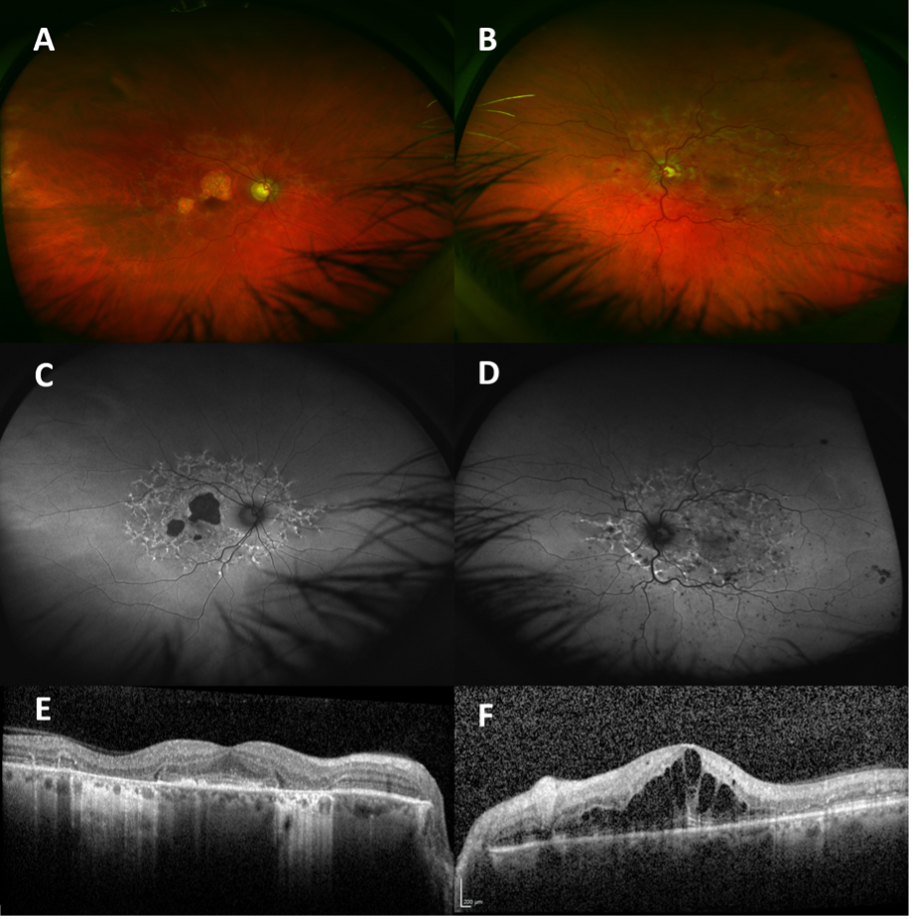
On examination, VA was 20/30 in the right eye and 20/200 in the left eye. He had early nuclear cataract and normal IOP in each eye. He had diffuse intraretinal hemorrhages, optic nerve head swelling, venous tortuosity, and macular thickening in the left eye. He had pisciform subretinal flecks extending to the vascular arcades with areas of GA in the right macula (Fig 2A, B). Autofluorescence demonstrated hyperautofluorescent and hypoautofluorescent areas consistent with current flecks and atrophic areas, respectively, most marked in the central macula of the right eye (Fig 2C, D). OCT confirmed areas of EZ atrophy, preservation of some foveal architecture in the right eye, and frank cystoid macular edema (CME) in the left eye (Fig 2E, F).
The clinical picture was consistent with subclinical Stargardt macular dystrophy (STGD1, OMIM #248200) with an acute overlay of left CRVO and CME. Genetic testing (as above) revealed compound heterozygous variants in the ABCA4 gene (c.6089G>A, p.(Arg2030Gln); c.4577C>T, p.(Thr1526Met)) confirming the STGD1 diagnosis. Treatment with intravitreal bevacizumab and later dexamethasone 0.7mg implant allowed resolution of CME and improvement of VA to 20/80 in the left eye with 3 years of follow-up. Glaucoma drainage implant surgery was required to lower IOP.
Discussion
These cases demonstrate that not all maculopathy in the elderly is AMD, particularly in those with high degrees of symmetry and specific fundus autofluorescence patterns.
IRDs typically present in childhood (e.g., Leber’s congenital amaurosis, STGD1) or early adulthood (e.g., retinitis pigmentosa), though some notable exceptions exist such as late-onset retinal degeneration (LORD, mean onset in 5th decade, caused by heterozygous C1QTNF5 variants). 8, 9
Pathogenic CRX variants (majority missense variants in exon 3) cause several phenotypes including autosomal dominant cone-rod dystrophy (~10%), classically manifesting a bulls-eye maculopathy with onset between 35 – 50 years.10-12 The age of onset in the present case is well beyond that typical of CRX-associated cone-rod dystrophy, even for the late-onset variant associated with entire CRX gene deletions (mean onset 51 years).12 STGD1 is typically associated with much earlier onset of disease (i.e., ABCA4 ~7-12 years old); however, a well-described late-onset (i.e., >45 years) phenotype has been described in association with less deleterious ABCA4 variants.13
Syndromic manifestations (e.g., sensorineural hearing loss in Usher syndrome or diabetes mellitus in Bardet-Beidl syndrome) in association with IRD and juvenile onset help to identify a genetic etiology, thus the combination of isolated macular atrophy and older adult symptom onset increases the likelihood of misdiagnosis as AMD.14
Fundus autofluorescence is of significant diagnostic benefit in cases of IRD, showing classic features in some conditions (e.g., dispersed subretinal flecks, macular atrophy, and peripapillary sparing in ABCA4-mediated STGD1).15 Visual electrophysiology is a useful screening test; however, it is not ubiquitously available as in the cases presented here. With increasing access to affordable high quality genetic testing, early genotyping may be an appropriate investigation early in the diagnostic pathway for clinically consistent cases with a high pre-test probability of the presence of an IRD.
In these cases, genetic testing is beneficial for the patient (e.g., to perhaps avoid unnecessary AREDS2 supplementation or complement inhibition treatment) and their family (e.g., reproductive planning and cascade screening of relatives).16Additionally, some IRD genotypes are associated with systemic comorbidities which may be amenable to prophylactic intervention. Any potential benefits of genotyping must be balanced with known treatment limitations and the potential for discovery of secondary or incidental findings (e.g., non-diagnostic carrier status, cancer risk alleles, etc.) dependent on the scope of the chosen genetic testing approach. 17, 18
Conclusion
An index of suspicion for non-AMD causes of macular disease in older adults should be maintained. Non-invasive and cost-effective screening tests such as fundus autofluorescence may help to identify those that could benefit from genetic testing to inform personal and family health decisions.
References
1. Liew G, Michaelides M and Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open 2014; 4: e004015. 2014/02/15. DOI: 10.1136/bmjopen-2013-004015.
2. You QS, Xu L, Wang YX, et al. Prevalence of retinitis pigmentosa in North China: the Beijing Eye Public Health Care Project. Acta Ophthalmol 2013; 91: e499-500. 20130613. DOI: 10.1111/aos.12163.
3. Whelan L, Dockery A, Wynne N, et al. Findings from a Genotyping Study of Over 1000 People with Inherited Retinal Disorders in Ireland. Genes 2020; 11 2020/01/23. DOI: 10.3390/genes11010105.
4. Neveling K, Collin RW, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012; 33: 963-972. 20120319. DOI: 10.1002/humu.22045.
5. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017; 390: 849-860. 2017/07/18. DOI: 10.1016/s0140-6736(17)31868-8.
6. Evans JR, Fletcher AE and Wormald RP. Causes of visual impairment in people aged 75 years and older in Britain: an add-on study to the MRC Trial of Assessment and Management of Older People in the Community. Br J Ophthalmol 2004; 88: 365-370. DOI: 10.1136/bjo.2003.019927.
7. McGwin G, Jr., MacLennan P and Owsley C. Association Between Pentosan Polysulfate Sodium and Retinal Disorders. JAMA ophthalmology 2022; 140: 37-42. DOI: 10.1001/jamaophthalmol.2021.4778.
8. Daich Varela M, Cabral de Guimaraes TA, Georgiou M and Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: current management and clinical trials. Br J Ophthalmol 2021 2021/03/14. DOI: 10.1136/bjophthalmol-2020-318483.
9. Lando L and Borooah S. Late-Onset Retinal Degeneration: Clinical Perspectives. Clin Ophthalmol 2022; 16: 3225-3246. 20220930. DOI: 10.2147/opth.S362691.
10. Sohocki MM, Sullivan LS, Mintz-Hittner HA, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet 1998; 63: 1307-1315. DOI: 10.1086/302101.
11. Hull S, Arno G, Plagnol V, et al. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Invest Ophthalmol Vis Sci 2014; 55: 6934-6944. 20140930. DOI: 10.1167/iovs.14-14715.
12. Yahya S, Smith CEL, Poulter JA, et al. Late-Onset Autosomal Dominant Macular Degeneration Caused by Deletion of the CRX Gene. Ophthalmology 2023; 130: 68-76. 20220805. DOI: 10.1016/j.ophtha.2022.07.023.
13. Lambertus S, Lindner M, Bax NM, et al. Progression of Late-Onset Stargardt Disease. Invest Ophthalmol Vis Sci 2016; 57: 5186-5191. DOI: 10.1167/iovs.16-19833.
14. Koenig R. Bardet-Biedl syndrome and Usher syndrome. Dev Ophthalmol 2003; 37: 126-140. 2003/07/25. DOI: 10.1159/000072043.
15. Varela MD, Laich Y, Hashem SA, et al. Prognostication in Stargardt disease using Fundus Autofluorescence: Improving Patient Care. Ophthalmology 2023 2023/06/19. DOI: 10.1016/j.ophtha.2023.06.010.
16. Heier JS, Lad EM, Holz FG, et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023; 402: 1434-1448. DOI: 10.1016/s0140-6736(23)01520-9.
17. Zhu J, Stephenson KAJ, Farrar GJ, et al. Management of significant secondary genetic findings in an ophthalmic genetics clinic. Eye (Lond) 2021 2021/05/05. DOI: 10.1038/s41433-021-01557-3.
18. Hanany M, Rivolta C and Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A 2020; 117: 2710-2716. 2020/01/23. DOI: 10.1073/pnas.1913179117.
Statement of Ethics
As per the Research Ethics Committee of the [redacted for review], case reports including ≤2 patients do not require ethics committee review or approval. (URL: redacted for review)
Conflict of Interest Statement
The authors declare no conflict of interest related to this case report.